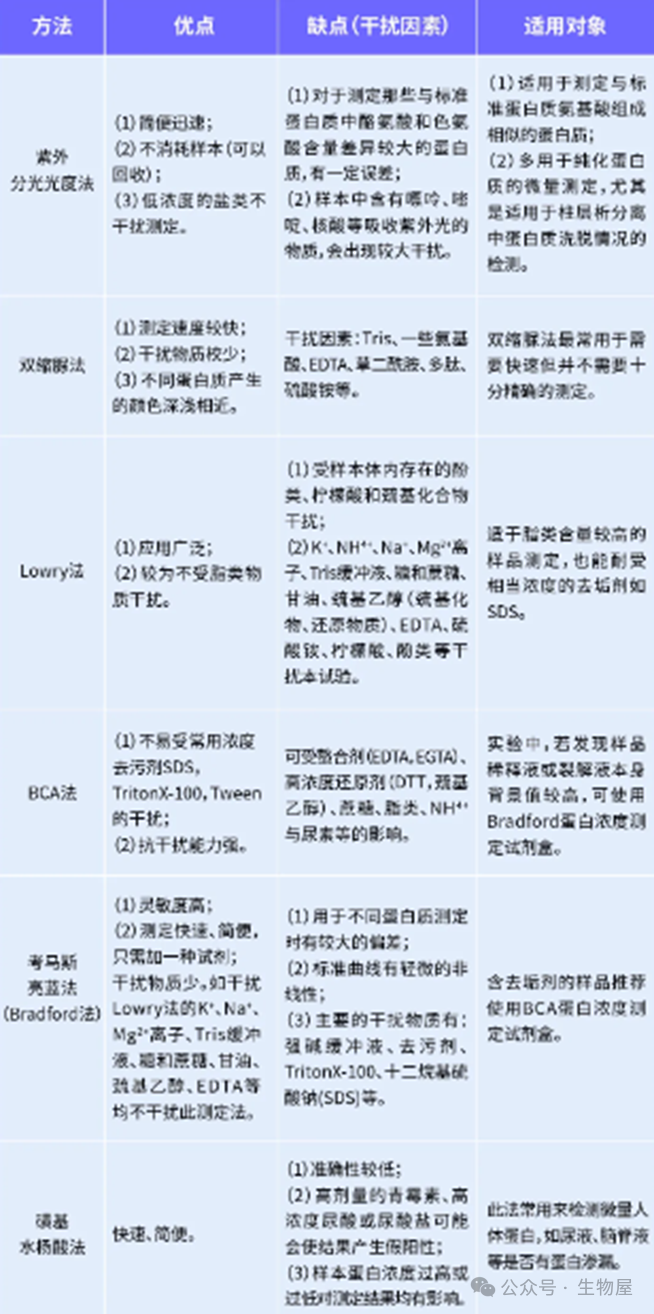
在生物化学和分子生物学研究中,准确测定蛋白质浓度是实验成功的关键之一。蛋白质是一种十分重要的生物大分子,其种类很多,结构多样,分子量相差也很大,因此建立一个比较通用或标准的定量方法是比较困难的。目前,测定蛋白的方法有多种,其优缺点也各不相同,因此,针对不同的目的,选择合适的蛋白测定方法尤为重要。
目前常用的测定蛋白浓度的方法主要包括:凯氏定氮法、紫外
-
分光光度法、双缩脲法、
Lowry
法、
Bradford
法、
BCA
法、磺基水杨酸法、荧光染料定量法、毛细管电泳、免疫测定法
(
如
ELISA)
。
经典的蛋白质含量测定方法。蛋白质是含氮的有机化合物。样品与浓硫酸和催化剂一同加热消化,使蛋白质分解,其中碳和氢被氧化为二氧化碳和水逸出,而样品中的有机氮转化为氨;氨与硫酸结合生成硫酸铵。加碱蒸馏使氨游离,用硼酸吸收后,再以硫酸或盐酸标准溶液滴定,根据酸的消耗量乘以换算系数
6.25(
如乳及乳制品换算系数为
6.38)
,即为蛋白质的含量。
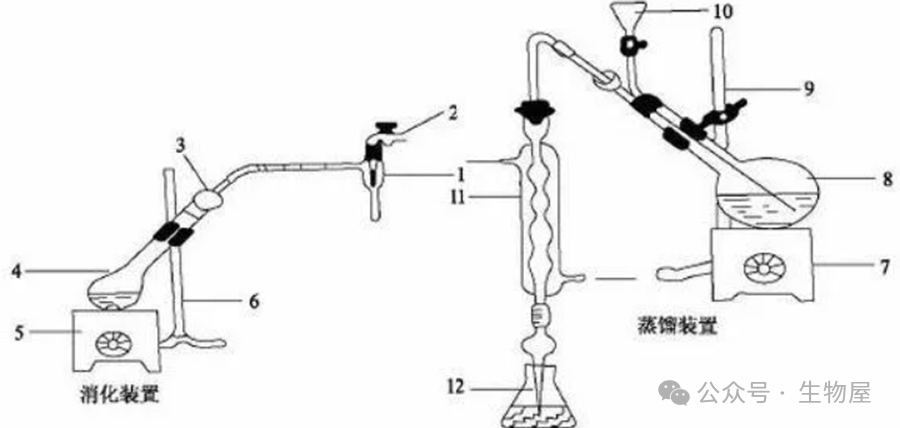
·
蛋白质的含氮量约为
16%
,即
1
克
(
蛋白质中的
)
氮相当于
6.25
克蛋白质,用凯氏定氮法测出的含氮量乘以
6.25
,即得样品中蛋白质的含量。
·
优点:
1.
测量范围广:可用于所有的蛋白质分析中;
2.
操作简单;
3.
测量浓度下限低:改进方法
(
微量凯氏定氮法
)
可测定样品中微量的蛋白质。
·
缺点:准确性不佳
——6.25
为含氮量换算为蛋白质含量的系数,这个系数来自蛋白质平均含氮量为
16
%,实际上各种蛋白质因氨基酸组成不同,含氮量不完全相同。

浓硫酸具有脱水性,使有机物脱水后被炭化为碳、氢、氮。浓硫酸又具有氧化性,可将有机物炭化后的碳氧化为二氧化碳,硫酸则被还原为二氧化硫。二氧化硫使氮还原为氨,本身则被氧化为三氧化硫,氨随之与硫酸作用生成硫酸铵留在酸性溶液中。
蛋白质
+13H₂SO₄→(NH₄)₂SO₄+6CO₂+12SO₂+16H₂O
消化时加入的硫酸铜是作催化剂,以加速分解反应,还可以加入氧化汞、氧化铜等作催化剂,为防止汞的污染,通常用硫酸铜较多。如果以汞或汞化合物作催化剂,则消化和加碱后,形成汞氨化合物。此化合物在蒸馏时不能完全分解,在这种情况下,必须加入锌粉或硫代硫酸钠或硫化钠,使汞氨化合物分解。
C+2CuSO₄→Cu₂SO₄+SO₂↑+CO₂↑
Cu₂SO₄+2H₂SO₄→2CuSO₄+2H₂O+SO₂
有机物消化完后,溶液具有清澈的硫酸铜的蓝绿色,同时硫酸铜在下一步蒸馏时可作碱性反应的指示剂。
在消化过程中添加硫酸钾,与硫酸反应生成硫酸氢钾,是为了提高溶液沸点,从而提高反应温度,加速反应过程。此外,也可以加硫酸钠、氯化钾等盐类来提高沸点。
在消化过程中,随着硫酸的不断分解、水分的不断蒸发,硫酸钾的浓度逐渐增大,沸点升高,加速了对有机物的分解作用。
加入过氧化氢,是利用其氧化性,以加快反应速度:
2H₂O₂→O₂+2H₂O
在消化完全的样品溶液中加入浓氢氧化钠使呈碱性,硫酸铵在碱性条件下释放出氨,通过加热蒸馏,氨随水蒸气蒸出:
(NH₄)₂SO₄+2NaOH →2NH₃+Na₂SO₄+2H₂O
加热蒸馏时放出的氨可用硼酸溶液进行吸收:
2NH₃+4H₃BO₃→(NH4)₂B₄O₇+5H₂O
待吸收完全后,用硫酸或盐酸标准溶液滴定生成的硼酸铵,属于盐类的滴定。硼酸为极弱的酸,在滴定中并不影响所用指示剂的变色反应。
(NH₄)₂B₄O₇+H₂SO₄+5H₂O→(NH₄)₂SO₄+4H₃BO₃
(NH₄)₂B₄O₇+2HCl+5H₂O→2NH₄Cl+4H₃BO₃
根据硫酸或盐酸溶液消耗的体积,计算总氮含量,再乘以蛋白质系数
6.25(
乳及乳制品换算系数为
6.38)
,即为粗蛋白的含量。
蛋白质分子中,酪氨酸、苯丙氨酸和色氨酸残基的苯环含有共轭双键,使蛋白质具有吸收紫外光的性质。吸收高峰在
280nm
处,其吸光度
(
即光密度值
)
与蛋白质含量成正比。此外,蛋白质溶液在
238nm
的光吸收值与肽键含量成正比。这一波长范围内的光吸收值与蛋白质浓度成正比,蛋白质浓度根据
Beer-Lambert
定律计算。
A
代表吸光度,
E
代表消光系数,
C
代表蛋白质浓度,
l
代表光径长度。消光系数可以根据蛋白质序列估测。
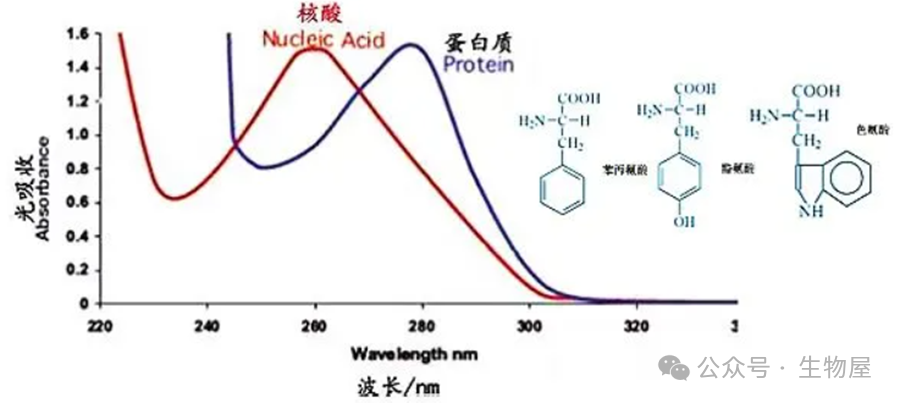
·
优点:
1.
简单快速;
2.
不消耗样品:检测过程中不对样品进行处理,故而测定后能回收样品。
·
缺点:
1.
普适性差:不同的蛋白质有不同的酪氨酸和色氨酸含量。对那些与标准蛋白质中酪氨酸和色氨酸含量差异大的蛋白质,有一定的误差。故要准确测量,必须要有待测蛋白质的纯品作为参考。
2.
曲线非线性:标准曲线也有轻微的非线性,因而不能用
Beer-Lambert
定律进行计算,而只能用标准曲线来测定未知蛋白质的浓度。
3.
干扰物质多:若样品中含有嘌呤、嘧啶及核酸等能吸收紫外光的物质,会出现较大的干扰。
·
另外,在使用此方法时,由于不同蛋白的序列不同,其所含有的酪氨酸和色氨酸所占总氨基酸的比例不同,因此造成了不同的蛋白具有不同的消光系数。在使用该方法进行测定蛋白浓度时,需要知道待测蛋白的消光系数,将通过紫外分光光度计测得的浓度除以蛋白的消光系数即为该蛋白的真实浓度。待测蛋白的消光系数可通过软件进行预测,在预测时只需要在网站中输入蛋白质的氨基酸序列即可给出相应的消光系数。预测蛋白消光系数的网址为:
https://web.expasy.org/protparam/
·
此外,进行紫外吸收法测定时,由于蛋白质吸收高峰常因
pH
的改变而有变化,因此要注意溶液的
pH
值,测定样品时的
pH
要与测定标准曲线的
pH
相一致。
1)
将待测蛋白质溶液倒入石英比色皿中,用配制蛋白质溶液的溶剂
(
水或缓冲液
)
作空白对照;
2)
在紫外分光度计上直接读取
280nm
的吸光度值
A280
;
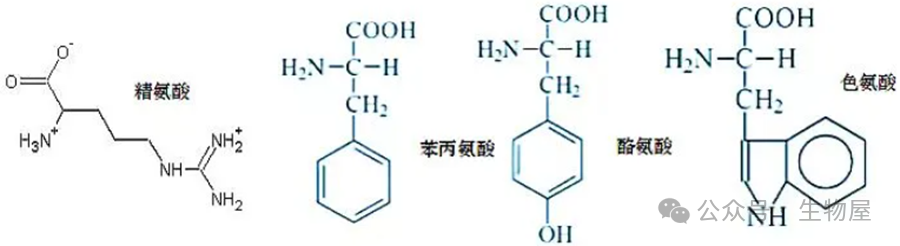
在酸性溶液中,考马斯亮蓝
G250
通过与蛋白质中的碱性氨基酸
(
精氨酸
)
和芳香族氨基酸残基相结合,使染料的最大吸收峰位置由
488nm
变为
595nm
,颜色从棕红色变为蓝色。结合到蛋白质分子上的染料数与蛋白所带正电荷成正比,通过颜色的强弱即可测定蛋白质浓度的高低,因此可以用该方法来对蛋白进行定量。
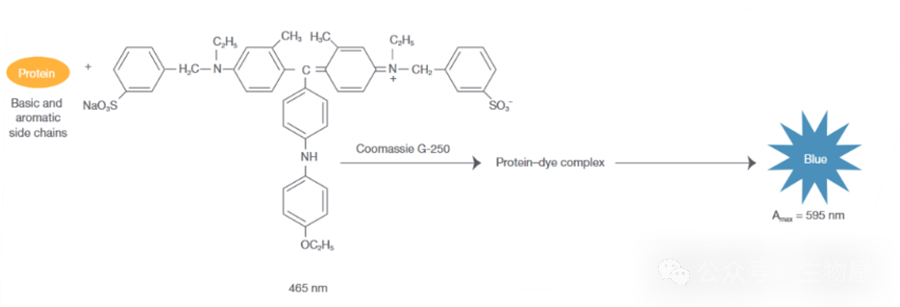
-
·因染料主要与蛋白质中精氨酸和芳香族氨基酸残基相结合,且两者在各种蛋白质含量不同,故用于不同蛋白质测定时有较大偏差,另外范德华力和疏水作用也会影响蛋白与染料的结合。
-
·Bradford
法测定蛋白浓度不受绝大部分样品中的化学物质的影响,如:
K⁺
、
Na⁺
、
Mg²⁺
离子、
Tris
缓冲液、糖和蔗糖、甘油、
EDTA
、
DTT
、
TCEP
和
β-
巯基乙醇等。但需要注意的是:由于高浓度洗涤剂会影响检测结果的可靠性,必须确保样品中的
SDS
的浓度低于
1
%,
Triton X-100
低于
0.1%
,
Tween-20
、
-60
、
-80
低于
0.06%
。
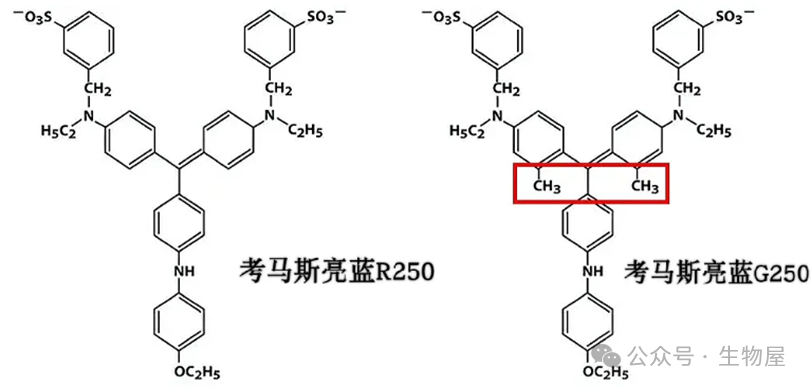
·检测蛋白浓度的考马斯亮蓝和染色SDS-PAGE胶的考马斯亮蓝不是同一种物质,前者采用考马斯亮蓝G250,后者采用考马斯亮蓝R250;G250和R250分子基本骨架一样,但是G250多了两个甲基,与蛋白反应迅速,染胶慢且脱色困难,故用于蛋白定量;R250与蛋白反应较缓慢,染胶较快且易于洗脱,故用于SDS-PAGE胶染色。
·Bradford法的优点是:
o
灵敏度高,据估计比
Lowry
法约高四倍,其最低蛋白质检测量可达
1mg
。这是因为蛋白质与染料结合后产生的颜色变化很大,蛋白质-染料复合物有更高的消光系数,因而光吸收值随蛋白质浓度的变化比
Lowry
法要大的多。
o
测定快速、简便,只需加一种试剂。完成一个样品的测定,只需要
5
分钟左右。由于染料与蛋白质结合的过程,大约只要
2
分钟即可完成,其颜色可以在
1
小时内保持稳定,且在
5-20
分钟之间,颜色的稳定性最好。因而完全不用像
Lowry
法那样费时和严格地控制时间。
o
干扰物质少。如干扰
Lowry
法的
K+
、
Na+
、
Mg2+
离子、
Tris
缓冲液、糖和蔗糖、甘油、巯基乙醇、
EDTA
等均不干扰此测定法。
o 由于各种蛋白质中的精氨酸和芳香族氨基酸的含量不同,因此Bradford法用于不同蛋白质测定时有较大的偏差,在制作标准曲线时通常选用g-球蛋白为标准蛋白质,以减少这方面的偏差。
o 仍有一些物质干扰此法的测定,主要的干扰物质有:去污剂、Triton X-100、十二烷基硫酸钠(SDS)和0.1 M NaOH。(如同0.1 M酸干扰Lowary法一样)。
o 标准曲线也有轻微的非线性,因而不能用Beer定律进行计算,而只能用标准曲线来测定未知蛋白质的浓度。
1)
取
16
支试管,
1
支作空白,
3
支留作未知样品,其余试管分为两组按表中顺序,分别加入样品、水和试剂,即用
1.0mg/ml
的标准蛋白质溶液给各试管分别加入:
0
、
0.01
、
0.02
、
0.04
、
0.06
、
0.08
、
0.1ml
,然后用无离子水补充到
0.1ml
。最后各试管中分别加入
5.0ml
考马斯亮兰
G250
试剂,每加完一管,立即在旋涡混合器上混合
(
注意不要太剧烈,以免产生大量气泡而难于消除
)
,未知样品加到新管中。
2)
加完试剂
2-5
分钟后,即可开始用比色皿,在分光光度计上测定各样品在
595nm
处的光吸收值
A595
,空白对照为第
1
号试管,即
0.1ml H2O
加
5.0ml G250
试剂。
注意:不可使用石英比色皿
(
因不易洗去染色
)
,可用塑料或玻璃比色皿,使用后立即用少量
95%
的乙醇荡洗,以洗去染色。塑料比色皿决不可用乙醇或丙酮长时间浸泡。
3)
用标准蛋白质量
(mg)
为横坐标,用吸光度值
A595
为纵坐标,作图,即得到一条标准曲线。由此标准曲线,根据测出的未知样品的
A595
值,即可查出未知样品的蛋白质含量。
双缩脲
(NH3CONHCONH3)
是两个分子脲经
180℃
左右加热,放出一个分子氨后得到的产物。在强碱性溶液中,双缩脲与
CuSO4
形成紫色络合物,称为双缩脲反应。凡具有两个酰胺基或两个直接连接的肽键,或能过一个中间碳原子相连的肽键,这类化合物都有双缩脲反应。
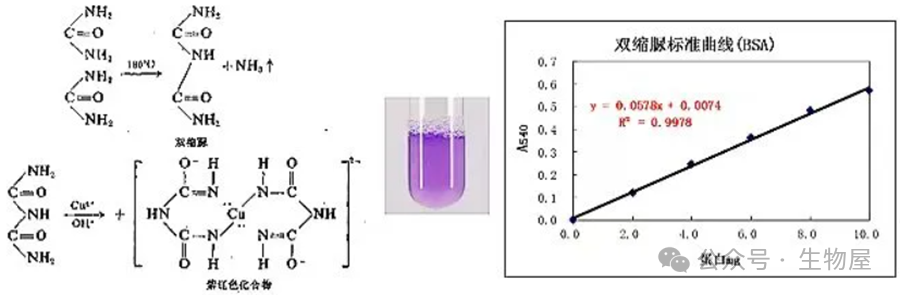
-
·紫色络合物颜色的深浅与蛋白质的含量成正比,而与蛋白质的相对分子质量及氨基酸成分无关,故可用来测定蛋白质含量。测定范围
1-10mg
蛋白质。

·此法的优点是较快速,不同的蛋白质产生颜色的深浅相近,以及干扰物质少。主要的缺点是灵敏度差。因此双缩脲法常用于需要快速,但并不需要十分精确的蛋白质测定。
·硫酸铵不干扰此呈色反应,使其有利于对蛋白质纯化早期步骤的测定。干扰此测定的物质包括在性质上是氨基酸或肽的缓冲剂,例如硫酸铵、Tris缓冲液和某些氨基酸等,因为它们给予阳性呈色反应。Cu²⁺也容易被还原,有时发现出现红色沉淀。
(1)
标准蛋白质溶液:用标准的结晶牛血清清蛋白
(bsa)
或标准酪蛋白,配制成
10mg/ml
的标准蛋白溶液,可用
bsa
浓度
1mg/ml
的
a280
为
0.66
来校正其纯度。如有需要,标准蛋白质还可预先用微量凯氏定氮法测定蛋白氮含量,计算出其纯度,再根据其纯度,称量配制成标准蛋白质溶液。牛血清清蛋白用
H2O
或
0.9%NaCl
配制,酪蛋白用
0
.
05NaOH
配制。
(2)
双缩脲试剂:称以
1.50
克硫酸铜
(CuSO4•5H2O)
和
6.0
克酒石酸钾钠
(KNaC4H4O6•4H2O)
,用
500
毫升水溶解,在搅拌下加入
300
毫升
10% NaOH
溶液,用水稀释到
1
升,贮存于塑料瓶中
(
或内壁涂以石蜡的瓶中
)
。此试剂可长期保存。若贮存瓶中有黑色沉淀出现,则需要重新配制。
2)
标准曲线的测定:
取
12
支试管分两组,分别加入
0
,
0.2
,
0.4
,
0.6
,
0.8
,
1.0
毫升的标准蛋白质溶液,用水补足到
1
毫升,然后加入
4
毫升双缩脲试剂。充分摇匀后,在室温
(20
~
25℃)
下放置
30
分钟,于
540nm
处进行比色测定。用未加蛋白质溶液的第一支试管作为空白对照液。取两组测定的平均值,以蛋白质的含量为横坐标,光吸收值为纵坐标绘制标准曲线。
3)
样品的测定:
取
2
~
3
个试管,用上述同样的方法,测定未知样品的蛋白质浓度。注意样品浓度不要超过
10mg/ml
。
5. Lowry
蛋白测定法
(Folin-
酚试剂法
)
用于蛋白质测定的
Lowry
反应是双缩脲方法的发展,第一步涉及到在碱性溶液中铜
-
蛋白质复合物的形成
(
蛋白质中的肽键和铜离子结合产生双缩脲反应
)
,
Folin-
酚试剂中的磷钼酸
-
磷钨酸试剂被铜
-
蛋白质复合物中蛋白质的酪氨酸和苯丙氨酸残基还原,产生深蓝色的钼蓝和钨蓝的混合物。在一定条件下,蓝色深度与蛋白的量成正比,由此可测定蛋白质的含量。
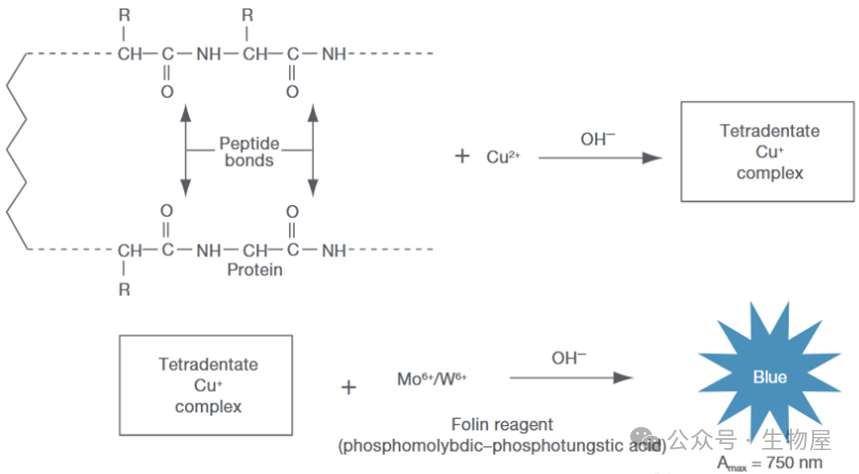
·这个测定法的优点是灵敏度高,比双缩脲法灵敏得多,缺点是费时间较长,要精确控制操作时间,标准曲线也不是严格的直线形式,且专一性较差,干扰物质较多。
·对双缩脲反应发生干扰的离子,同样容易干扰Lowry反应。而且对后者的影响还要大得多。酚类、柠檬酸、硫酸铵、Tris缓冲液、甘氨酸、糖类、甘油等均有干扰作用。浓度较低的尿素(0.5%),硫酸纳(1%),硝酸纳(1%),三氯乙酸(0.5%),乙醇(5%),乙醚(5%),丙酮(0.5%)等溶液对显色无影响,但这些物质浓度高时,必须作校正曲线。含硫酸铵的溶液,只须加浓碳酸钠―氢氧化钠溶液,即可显色测定。若样品酸度较高,显色后会色浅,则必须提高碳酸钠―氢氧化钠溶液的浓度1~2倍。
·进行测定时,加folin―酚试剂时要特别小心,因为该试剂仅在酸性pH条件下稳定,但上述还原反应只在pH=10的情况下发生,故当folin一酚试剂加到碱性的铜―蛋白质溶液中时,必须立即混匀,以便在磷钼酸―磷钨酸试剂被破坏之前,还原反应即能发生。
·此法也适用于酪氨酸和色氨酸的定量测定。
·可检测的最低蛋白质量达5mg。通常测定范围是20~250mg。
1)
标准曲线的测定:
取
16
支大试管,
1
支作空白,
3
支留作未知样品,其余试管分成两组,分别加入
0
,
0.1
,
0.2
,
0.4
,
0.6
,
0.8
,
1.0
毫升标准蛋白质溶液
(
浓度为
250mg/ml)
。用水补足到
1.0
毫升,然后每支试管加入
5
毫升试剂甲,在旋涡混合器上迅速混合,于室温
(20
~
25℃)
放置
10
分钟。再逐管加入
0.5
毫升试剂乙
(folin―
酚试剂
)
,同样立即混匀。这一步混合速度要快,否则会使显色程度减弱。然后在室温下放置
30
分钟,以未加蛋白质溶液的第一支试管作为空白对照,于
700nm
处测定各管中溶液的吸光度值。以蛋白质的量为横坐标,吸光度值为纵坐标,绘制出标准曲线。
2)
样品的测定:
取
1
毫升样品溶液
(
其中约含蛋白质
20~250
微克
)
,按上述方法进行操作,取
1
毫升蒸馏水代替样品作为空白对照。通常样品的测定也可与标准曲线的测定放在一起,同时进行。
3)
根据所测样品的吸光度值,在标准曲线上查出相应的蛋白质量,从而计算出样品溶液的蛋白质浓度。
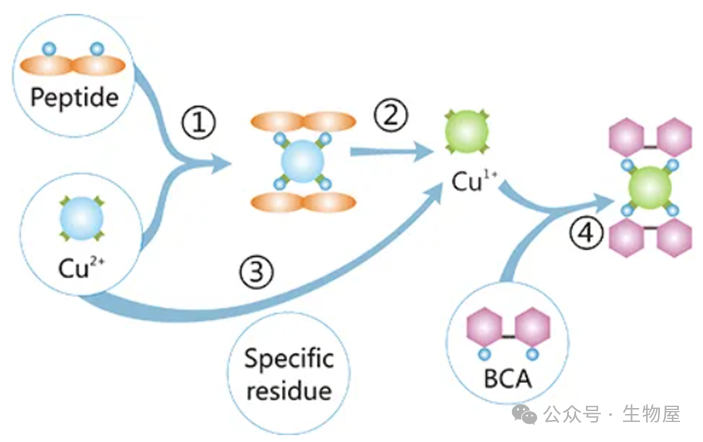
改良的
Lowry
法,使用二喹啉酸
(bicinchoninic acid)
作为反应剂。本方法将双缩脲反应和显色反应结合在一起:前者为在碱性介质中,蛋白质可将
Cu2+
还原成
Cu1+
的反应,后者为使用含有二喹啉甲酸
(BCA)
的独特试剂,利用比色法检测
Cu1+
,具有高灵敏度和高选择性的特点。此检测方法中产生的紫色显色物质是由两分子的
BCA
和一分子的亚铜离子螯合而形成的。
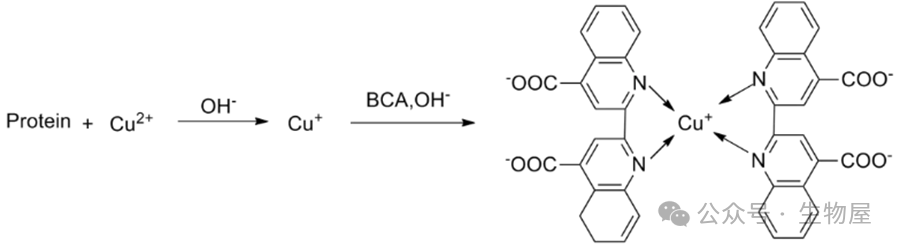
·优点: 在碱性溶液中BCA 比Folin试剂稳定,操作简便仅需一种试剂即可完成检测。
o
1
)灵敏度高,检测浓度下限达到
10μg/ml
,最小检测蛋白量达到
0.2μg
,待测样品体积为
1-20μl
;
o
2
)测定蛋白浓度不受绝大部分样品中的去污剂等化学物质的影响,可以兼容样品中高达
5%
的
SDS
,
5%
的
Triton X-100
,
5%
的
Tween 20
,
60
,
80
;
o
3
)在
20-1000μg/ml
浓度范围内有良好的线性关系;
o
4
)检测不同蛋白质分子的变异系数远小于考马斯亮蓝法蛋白定量;
o
5
)显色速度快:相同的样品孵育较短时间即可进行吸光度测定。
·缺点:容易受到能与铜离子反应的螯合剂,例如EDTA,还原性试剂(β-巯基乙醇)以及蛋白质内半胱氨酸等的影响。通常建议样品中的EDTA含量小于10mM,DTT含量小于1mM,巯基乙醇含量低于1mM。
室温完全溶解蛋白标准品
,
取
20µl 5mg/ml BSA
蛋白标准溶液用
PBS
溶液稀释至
100µl,
使其终浓度为
1.0 mg/ml
。
将
A
液与
B
液按照
50:1
的比例混合,配成
BCA
工作液,并混匀。注:此处一定要精准计算待测孔所用工作液的量,如测定
4
个蛋白样本,均做
3
个复孔,那么加上标曲一共有
39
孔,每个测定孔均需加入
200µl
的工作液,那么共需
7800µl
工作液,则
A
液:
B
液
=7800:156
。
按照第二步所示表格加标准曲线测定孔,将待测样本以适当体积加入并用
PBS
补足至
20ul(
待测样本加入的量可按照经验值进行估计
)
。
向各孔中加入
200µl
的工作液,混匀后
37℃
孵育
30min
,亦可室温放置
2h
。
测定
562nm
处的吸光度,记录数值,代入标曲计算浓度。
a.
新建一个
Excel
表格,将得到的标准品的吸光度按照下方表格排列,并按照试剂说明说输入相应标准品的浓度;
b.
选中标准品的吸光度和浓度,点击
“
插入
”
,选择散点图;
c.
点击散点图上的任意一点,右键选择添加趋势线;点击设置,趋势线选项选择
“
线性
”
,勾选
“
显示公式
”
和
“
显示
R
平方值
”
,即可得到标准曲线;
注:横坐标为吸光度,纵坐标为浓度,R2需要大于0.99方可使用。
d.
计算样本浓度:代入公式直接计算即可,但要记得样本稀释倍数再回算回去。
1)
标准品可现配现用,也可配制后在
-20℃
保存一年,但工作液需要现配现用。
2)
为保证蛋白浓度测量的准确性,最好做
3
个复孔,必要时剔除离群值。
3) BCA
法测蛋白浓度易受时间及温度的影响,所以最好每次测定都做标准曲线。
4)
在制作标曲时,一定要分清横纵坐标哪个代表浓度哪个代表吸光度,不要搞错。
5)
加样时一定要准确加样且尽可能避免气泡产生,以免影响测量
(
加样结束可以用注射器戳破气泡,并将
96
孔板放在摇床上混匀片刻,以使样品与工作液充分接触
)
。
6)
孵育结束应尽快检测吸光度,并尽可能加快检测速度,以免带来误差。
7)
一般情况下,样品的蛋白浓度是比较高的,需要用
PBS
溶液进行稀释后测量,稀释倍数可根据经验值进行调整,保证浓度测量在标准曲线范围内进行。
NanoOrange
是一种新型的用于蛋白质精确定量的荧光染料。当
NanoOrange
处于溶液状态时没有荧光活性,与蛋白作用后,在
470-490nm
处激发,产生
570-590nm
的发射光,发光的强度与蛋白质的含量具有一定的线性,可以使用荧光计对发光强度进行检测,根据标曲计算蛋白的含量。

·
使用
NanoOrange
最低可以检测到
100ng/ml
的蛋白样品,其灵敏度高于
Bradford
法和
Lowry
法等分光光度方法,也高于
280nm
吸光光度法。
·
与
Bradford
法比较,
NanoOrange
受不同蛋白影响更小。
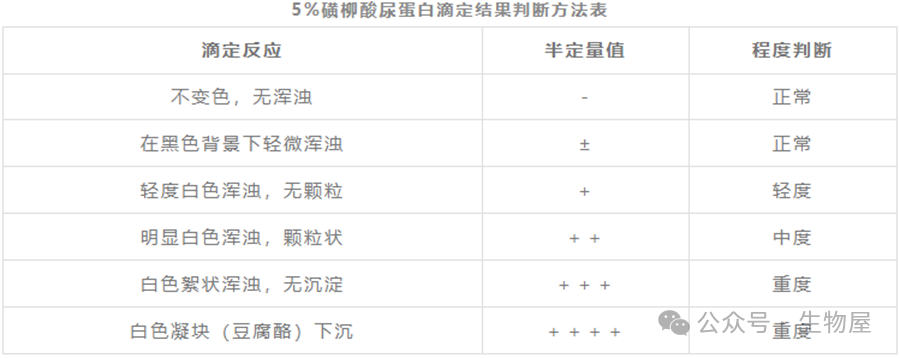
磺基水杨酸法(
SSA
),又称为磺柳酸法。蛋白质为两性物质,在酸性环境中带正电荷,而磺基水杨酸根带负电。在略低于蛋白质等电点的酸性环境下,磺基水杨酸根离子与蛋白质氨基酸阳离子结合,形成不溶性蛋白盐而沉淀,判定为阳性。沉淀的量或者溶液反应后的浑浊程度,可以反映蛋白质的含量,是尿液蛋白质的定性或者半定量检查方法。
·
优点是反应灵敏、操作简便、结果显示快,且能够与白蛋白、糖蛋白、球蛋白和本周蛋白等发生反应,特别适合基层检验科尿液蛋白的复检方法。
·
缺点是准确性较低;高剂量的青霉素、高浓度尿酸或尿酸盐可能会使结果产生假阳性;样本蛋白浓度过高或过低对测定结果均有影响。
·
此法常用来检测微量人体蛋白,如尿液、脑脊液等是否有蛋白渗漏。
·
该方法的敏感度达到
50mg/L
,因而有一定的假阳性。

9.
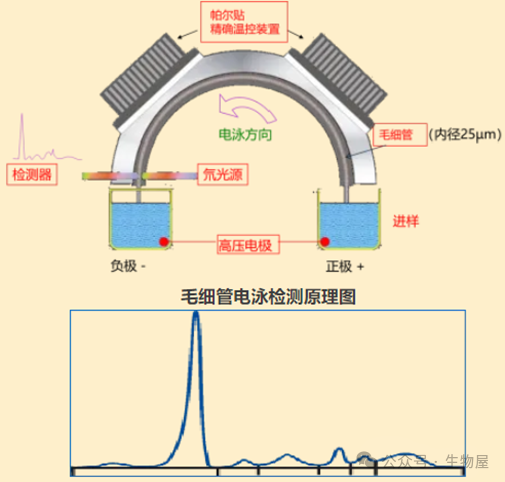
毛细管电泳
(Capillary electrophoresis, CE)
利用蛋白质的等电点和分子大小的不同,在同一
pH
值缓冲液中所带电荷的差异性,在电场中泳动速度和方向不同,使不同的蛋白质分子具有不同的电泳迁移率,从而把蛋白分为不同的区带,然后不同的区带进行定量分析。
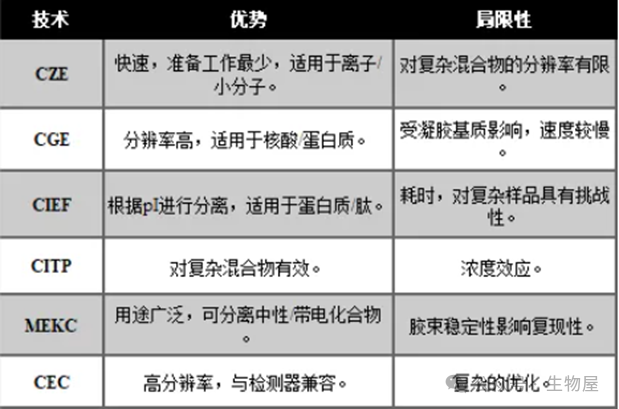
毛细管电分离包含一系列具有不同特性的技术,可适应不同的分离或样品要求。每种
CE
技术都提供了一种特异性的分析带电分子的方法,可满足特定的分析物需求。选择合适的技术需要在速度、分辨率、兼容性和复杂性之间进行权衡。
毛细管区带电泳(
CZE
)是最常用的
CE
技术,其基本工作原理已在上一节中说明。毛细管区带电泳的特点是样品制备要求低、速度快。该技术特别适用于离子和小分子,但在处理复杂混合物时,其分辨率可能会受到限制,因为密切相关的物质可能会洗脱出宽阔的峰值。
毛细管凝胶电泳通过在毛细管中加入筛分基质(通常是交联聚合物)来提高分离效果。这种基质比小分子更能阻止大分子的迁移,从而实现高分辨率分离。
CGE
尤其适用于核酸和蛋白质的分离,可对分子量差异进行精确分析。然而,
CGE
对凝胶基质的依赖使其与其他技术相比速度较慢。
该技术利用分析物等电点(
pI
)的差异实现分离。毛细管中充满两性缓冲液,形成
pH
梯度。分析物迁移到各自的等电点(
pI
)处,在此变成电中性而停止。这种技术非常适合具有不同
pI
的蛋白质和肽。不过,由于需要校准
pI
,
CIEF
比较耗时,而且复杂的样品会对分辨率提出挑战。
在
CITP
中,分析物的分离是以离子迁移率为基础逐步进行的。迁移率较低的前导电解质和迁移率较高的后导电解质将分析物夹在中间,并将它们推向检测器。
CITP
在分离复杂混合物方面表现出色,可确保每个分析物占据一个独特的迁移区域。然而,浓度效应会导致峰形失真。
微束电动毛细管色谱是一种融合了电泳和色谱的混合技术。它是在电解质中加入表面活性剂胶束。中性分析物在水相和胶束核心之间分配,而带电分析物则根据其电荷质量比迁移。
MEKC
的多功能性使其既能分离中性化合物,也能分离带电化合物。不过,胶束的稳定性会影响复现性
。
毛细管电色谱结合了色谱和电泳原理。固定相覆盖在毛细管内壁上,与分析物发生类似于传统色谱法的相互作用。这种相互作用与电泳流动性相结合,实现了高分辨率分离。
CEC
与各种检测方法兼容,能够分离复杂的样品。不过,固定相和流动相的优化可能会很复杂
。
3)
分析电泳图谱,根据迁移时间和峰面积计算蛋白质浓度。
ELISA
技术基于抗原或抗体的固相化及抗原或抗体的酶标记。将抗原或抗体结合在固相载体表面并保持其免疫学活性,此外将抗原或抗体进行酶标记,使其既保留其免疫学活性,又保留酶的活性。在测定时,待测样品
(
含有要检测含量的抗体或抗原
)
与固相载体表面的抗原或抗体发生抗原抗体反应,特异性结合形成复合物。之后通过洗涤的方法使固相载体上形成的抗原抗体复合物与液体中未反应的其他物质分离。再加入酶标记的抗原或抗体,通过抗原抗体反应而结合在固相载体上。此时固相上的含酶量与样品中待检物质的量呈正比。加入酶反应的底物后,底物被酶催化成为有色产物,产物的量与样品中待检物质的量直接相关,故可根据呈色的深浅进行定性或定量分析。由于酶的催化效率很高,间接地放大了免疫反应的结果,使测定方法达到很高的敏感度。
在实际应用中,通过不同的设计,具体的方法步骤可有多种。即:用于检测抗体的间接法、用于检测抗原的双抗体夹心法以及用于检测小分子抗原或半抗原的抗原竞争法等。
将抗原直接固定在固相载体上,加入酶标记的一级抗体,即可测定抗原总量,此一级抗体的特异性非常重要。
优势:操作手续简短,因无须使用二抗可避免交互反应。
缺点:试验中的一抗都得用酶标记,但不是每种抗体都适合做标记,费用相对提高。
此测定方法与直接法类似,差别在于一级抗体没有酶标记,改用酶标记的二级抗体去辨识一级抗体来测定抗原量。
优势:二抗可以加强信号,而且有多种选择能做不同的测定分析。不加酶标记的一级抗体则能保留它更多的免疫反应性。
3)
双抗体夹心法
(sandwich ELISA)
被检测的抗原包被在两个抗体之间,其中一个抗体将抗原固定于固相载体上,即捕捉抗体。另一个则是检测抗体,此抗体可用酶标记后直接测定抗原的量;或不标记,再透过酶标记的二级抗体来测定抗原的量。这两种抗体必须小心选取,才可避免交互反应或竞争相同的抗原结合部位。
4)
竞争法
(competitive ELISA)
样本里的抗原
(
自由抗原
)
和纯化并固定在固相载体上的抗原
(
固定抗原
)
一起竞争相同的抗体,当样品里的自由抗原越多,就可以结合越多的抗体,而固定抗原就只能结合到较少的抗体,反之亦然。经清洗步骤,洗去自由抗原和抗体的复合物,只留下固定抗原和抗体的复合物,拿来与只有固定抗原的对照组结果相比较,根据呈色差异就可计算出样品里的抗原含量。
5)
新
ELISA
技术:基于细胞法
(cell-based ELISA)
是一种新的定性蛋白检测技术,将细胞直接在微孔板里培养,待检测时,不需抽提蛋白和裂解细胞,便可直接测量微孔板里蛋白经刺激或抑制作用后的变化。
优势:无需裂解细胞,所以目标蛋白损失少,可测定完整细胞、黏附细胞、还有非粘附细胞。
1)
加样:
加一定稀释的待检样品
100 μL
于上述已包被之反应孔中,置
37℃
孵育
1h
。然后洗涤。
(
同时做空白孔,阴性对照孔及阳性对照孔
)
。
2)
加酶标抗体:
于各反应孔中,加入新鲜稀释的酶标抗体
(
按照说明书进行稀释
)100 μL
。
37℃
孵育
0.5
~
1h
,洗涤
3
次。
3)
加底物液显色:
于各反应孔中加入临时配制的
TMB
底物溶液
100 μL
,
37℃10
~
30min
。加入终止液后
30 min
内,使用酶标仪测量
450nm
的吸光度值,设定
540nm
或
570nm
作为校正波长。如果没有使用双波长校正,结果准确度可能会受影响;
4)
计算结果:
将每个标准品和样品的校正吸光度值
(OD450-OD540/OD570)
、复孔读数取平均值,然后减去平均零标准品
OD
值。使用计算机软件作四参数逻辑
(4-PL)
曲线拟合创建标准曲线。另一种方法是,可以通过绘制标准品浓度做对数与相应
OD
值对数生成曲线,并通过回归分析确定最佳拟合线。这个过程可生成一个足够使用但不太精确的数据拟合。

3.
Extinction coefficient-a guide to understanding extinction coefficients, with emphasis on spectrophotometric determination of protein concentration, Thermo scientific, TR0006.4.
4.
https://bio-protocol.org/cn
5. https://star-protocols.cell.com/search
6. Advances in capillary electrophoresis for the life sciences. J Chromatogr B. 2019; 1118-1119: 116-136. doi:10.1016/j.jchromb.2019.04.020

